L’altra facia della soia (e della quinoa)
Testo di Rosanna Novara Topino
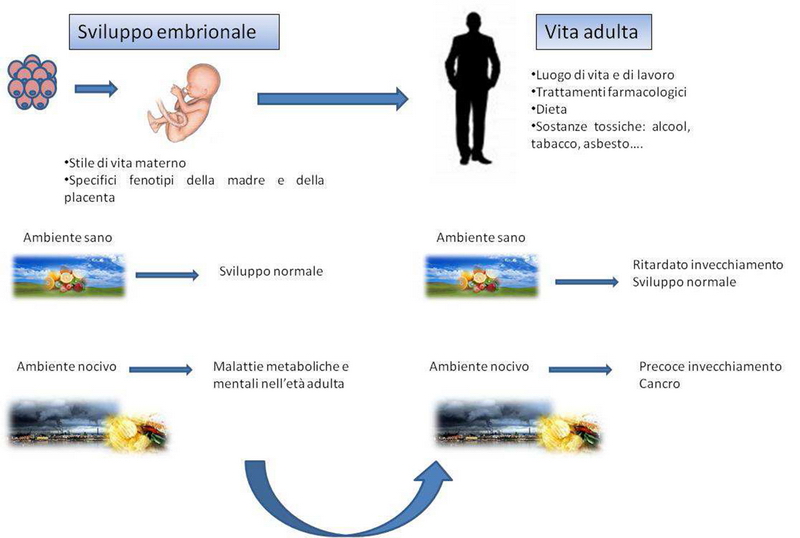
La sua dentatura dimostra che l’essere umano è onnivoro. Anche per questo è riuscito ad adattarsi all’ambiente. Oggi il problema è trovare una dieta che risponda alle esigenze nutritive e, allo stesso tempo, rispetti gli animali e l’ambiente. I pro e i contro della scelta vegetariana e di quella vegana.
Come dimostra la nostra dentatura, in cui sono contemporaneamente presenti sia canini di modeste dimensioni, ma comunque atti a mangiare carne, che denti molari e premolari per triturare vegetali (mentre gli incisivi servono per mordere ogni tipo di cibo), l’essere umano è onnivoro e forse questa caratteristica ha determinato il successo evolutivo dell’Homo sapiens, per la sua grande capacità di adattamento all’ambiente e a tutti i tipi di cibo in esso presenti. Tuttavia non siamo erbivori, cioè non disponiamo del corredo enzimatico necessario per ricavare energia dalla cellulosa, lo zucchero complesso maggiormente presente nei vegetali. Per non andare incontro a carenze vitaminiche, soprattutto di vitamina B12 e di aminoacidi essenziali, presenti nella carne e che non siamo capaci di sintetizzare in proprio, siamo quindi costretti a mangiare di tanto in tanto cibi di origine animale. In pratica, ciò che dobbiamo introdurre nel nostro organismo con l’alimentazione è strettamente correlato al nostro Dna, ereditato dai nostri antenati, che in effetti si cibavano anche, ma non solo, di carne.

Vegetariani e vegani: non va tutto bene
Resta il dilemma del rispetto degli animali. Da questo punto di vista la dieta che meglio concilia quest’ultimo con le nostre esigenze nutritive è quella latte-ovo-vegetariana, anche se si rischia di eccedere nel consumo di latte e uova, per ottenere la stessa qualità di principi alimentari presenti in quantità superiore nella carne. Inoltre, questa dieta è inadatta per gli intolleranti al lattosio e per coloro che hanno problemi di ipercolesterolemia familiare.
La dieta vegana, nata come scelta etica del rispetto per gli animali e per l’ambiente, esclude invece totalmente i prodotti di origine animale e li sostituisce con prodotti vegetali particolarmente ricchi di proteine come la soia (abbondantemente utilizzata nella cucina vegana per produrre alimenti che ricordano, per consistenza, la carne e i prodotti caseari senza avere però le stesse proprietà nutritive), la quinoa, le mandorle, l’avocado e gli anacardi. In realtà questa dieta è controversa perché, per ricavare le proteine necessarie, utilizza grandi quantità di soia, con il grande impatto ambientale che essa comporta (disboscamenti, impoverimento dei suoli, ecc.).
Un altro alimento largamente utilizzato nella cucina vegana (ma non solo) è la quinoa coltivata in Bolivia, Cile, Perù ed Ecuador. Si tratta di una pianta erbacea della famiglia delle Chenopodiacee (a cui appartengono anche spinaci e barbabietole), dalle grandi proprietà nutritive perché composta per il 60% da amido e per il 12-18% da proteine ricche di due aminoacidi essenziali: la lisina e la metionina. La lisina è necessaria allo sviluppo e alla fissazione del calcio sulle ossa e inoltre favorisce la produzione di anticorpi, ormoni ed enzimi. La metionina ha un’azione lipolitica e partecipa ai processi di detossificazione e di eliminazione dei prodotti metabolici di scarto.
Nel 2013 la quinoa è stata dichiarata «cibo dell’anno» dall’allora segretario delle Nazioni Unite Ban Ki-moon e la sua produzione è stata interessata da un vero e proprio boom, vista la domanda a livello mondiale. In Bolivia, i terreni destinati alla produzione di questa pianta sono passati, nel giro di pochi anni, da 10.000 a 50.000 ettari e il 90% dei semi prodotti è destinata all’esportazione. In pratica i terreni, che in passato producevano una grande quantità di colture diverse, si sono trasformati in monocolture di quinoa. Data la grande richiesta, il prezzo della quinoa è aumentato fino a triplicare, al punto che per i contadini del Perù, per i quali questa pianta è sempre stata parte della cucina tradizionale da migliaia di anni, è diventato impossibile cibarsene. In Bolivia il suo prezzo è diventato quattro volte tanto quello del riso o di altri cereali. La quinoa viene perciò venduta quasi del tutto o scambiata con Coca-Cola, dolciumi e cibo della dieta occidentale. Anche per questa causa, attualmente il 19,5% dei bambini peruviani (dato Unicef) soffre di malnutrizione. Inoltre, in passato la quinoa veniva coltivata solo sui pendii delle Ande, mentre i terreni più a valle erano destinati all’allevamento di lama e alpaca. Ora molti di questi terreni sono utilizzati per la coltivazione della pianta, mentre gli allevamenti si sono notevolmente ridotti e sono stati confinati nelle zone collinari, riducendo ulteriormente le possibilità di sostentamento del popolo andino. A tutto questo si aggiunge l’uso di fertilizzanti e di anticrittogamici di scarsa qualità, che inquinano il suolo, le falde acquifere e l’aria, impoverendo il terreno, la cui resa sta diminuendo progressivamente.
Altro alimento controverso presente spesso nella dieta vegana sono gli anacardi coltivati per il 40% nel Vietnam, spesso da tossicodipendenti condannati ai lavori forzati in centri di recupero, mentre il restante viene prodotto nelle zone più povere della Costa d’Avorio e dell’India. Qui gli anacardi vengono ripuliti dai loro gusci a mani nude dalle donne, che non possono permettersi i guanti per proteggersi dall’olio caustico formato dagli acidi anacardici cardolo e metilcardolo, i quali provocano ferite permanenti simili a ustioni sulla pelle.
La dieta mediterranea
È evidente che la dieta vegana è molto rispettosa degli animali, ma non altrettanto delle persone che producono parte del cibo utilizzato. Questa dieta sarebbe forse più rispettosa sia degli animali che degli umani, se utilizzasse esclusivamente frutta e verdura, cereali e oleaginose a Km 0. Resta il fatto che necessita dell’integrazione di vitamine del gruppo B, in particolare di B12, per non incorrere nel rischio di anemia perniciosa e di disturbi neurologici ed è decisamente sconsigliata per i bambini, gli adolescenti, le donne in gravidanza e in allattamento.
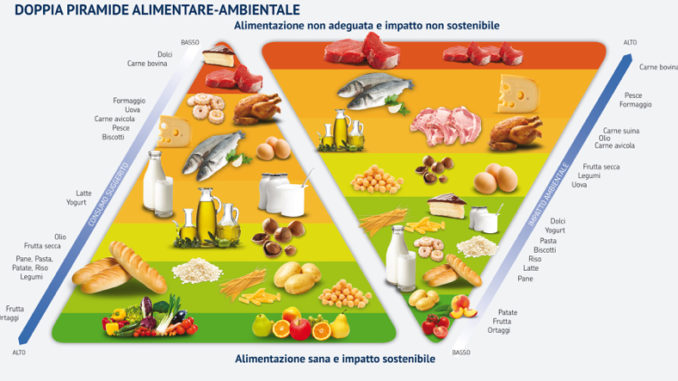
Per chi non può fare a meno della carne, la dieta più rispettosa dell’ambiente è quella mediterranea, che prevede un abbondante consumo di cereali, di ortaggi e frutta, un consumo medio di pesce e un limitato consumo di carne e di latticini, mentre gli zuccheri semplici sono ridotti al minimo indispensabile.

I gas serra delle risaie
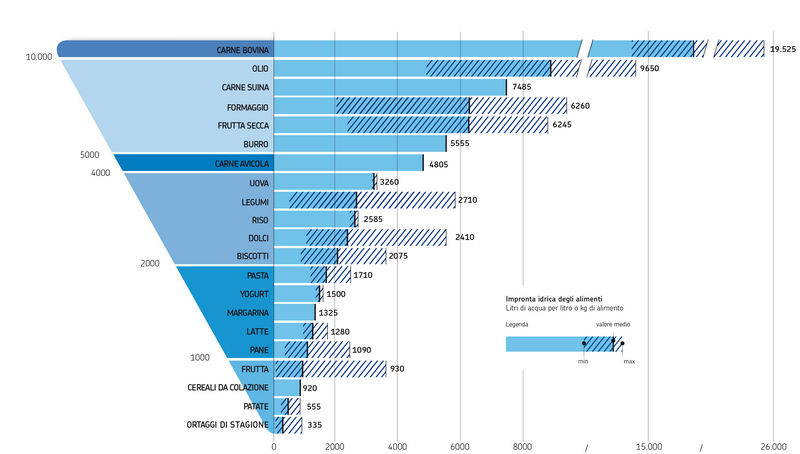
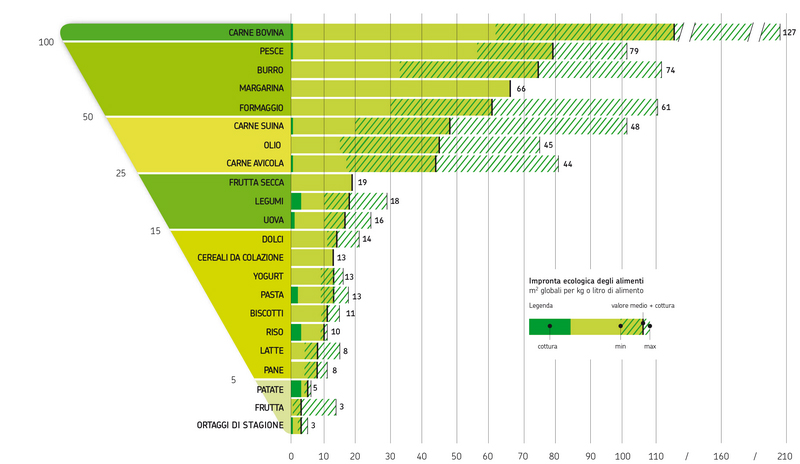
Sia la dieta vegetariana che quella vegana, se particolarmente ricche di riso possono avere un certo peso a livello di impatto ambientale. Questo perché, come si è visto da recenti studi, tra i principali produttori di gas serra al mondo ci sono le risaie a sommersione, che emettono metano e protossido di azoto in quantità tali da potere essere paragonate all’attività di almeno 200 centrali a carbone per quanto riguarda il loro effetto sul riscaldamento globale, ma la stima è per difetto, se si considerano le emissioni sul lungo periodo. Perché avviene tutto ciò? Il riso, almeno nel 75% dei casi, viene coltivato in sommersione, tecnica che soddisfa sia le esigenze idriche che quelle di termoregolazione delle piante, con limitazione delle escursioni termiche a cui esse sarebbero esposte se coltivate su terreno asciutto. Questo però comporta che si crei un ambiente anaerobico sommerso con la crescita di batteri anaerobi metanogeni, che riescono a ottenere metano dalla digestione dell’amido presente nelle radici delle piante, liberandolo nell’atmosfera. Per quanto riguarda la produzione di protossido di azoto, essa deriva dalla nitrificazione e denitrificazione del suolo, a seguito dell’uso dei fertilizzanti azotati. Considerando l’estensione delle risaie sulla Terra, le emissioni di metano ad esse dovuto rappresentano il 20% del totale. Tali emissioni possono variare a seconda del clima, dell’annata e del modo di coltivare il riso. Mediamente per la coltura in sommersione continua vengono emessi 185 Kg/ha/anno, mentre per la semina interrata con sommersione differita vengono rilasciati 115 Kg/ha/anno e infine 5Kg/ha/anno per la semina a irrigazione turnata, che però rende il 40% in meno come prodotto. Il rovescio della medaglia è che man mano che diminuisce la produzione di metano delle risaie, aumenta quella di protossido d’azoto, gas serra 12 volte più potente del metano stesso e 296 volte più della CO2, come già detto. Nelle risaie a sommersione continua ne viene rilasciato 1 Kg/ha/anno, per la semina interrata a sommersione differita 1,6 Kg/ha/anno e per quella a irrigazione turnata 4,5 Kg/ha/anno. Se tutte le risaie del mondo fossero convertite a irrigazione turnata, le emissioni annue di metano diminuirebbero di 12 Tg, ma occorrerebbe detrarre l’incremento di protossido di azoto stimabile in 7,7 Tg di metano equivalente. Questo però comporterebbe la riduzione del 30% della produzione mondiale di riso, che è la seconda coltura più importante per la nutrizione umana dopo quella del frumento. Ne varrebbe la pena? La risposta è ardua, perché altri studi hanno dimostrato che elevati livelli di CO2 causerebbero una riduzione dei tassi di minerali, di proteine e di vitamine di questo cereale, vale a dire che i gas serra stanno anche rendendo il riso meno nutriente.
Nel nostro piccolo
Quello che possiamo fare noi, per pesare meno sull’ambiente con la nostra alimentazione è probabilmente fare sempre attenzione alla provenienza dei cibi, prediligendo quelli a Km 0, o addirittura coltivandoci qualche ortaggio e frutta direttamente in proprio, se possibile (per pomodori e insalata bastano un paio di cassette di terra sul balcone), in modo da non favorire più di tanto il trasporto su gomma.
Rosanna Novara Topino
(fine seconda parte – continua)



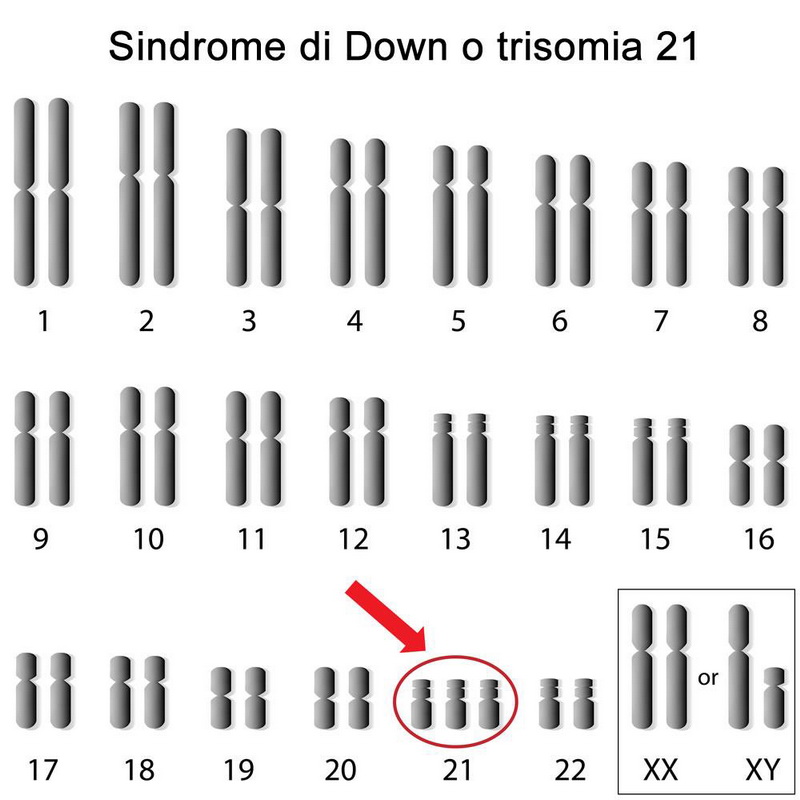
 Secondo l’Onu, l’Italia, che conta oltre 4 milioni di disabili (si tratta di un valore stimato, poiché non ci sono dati certi a riguardo), non è un paese a misura di disabile per diverse ragioni tra cui l’esiguità dei fondi messi a disposizione, il clima discriminatorio, le barriere architettoniche, le disuguaglianze in campo occupazionale, sanitario e scolastico.
Secondo l’Onu, l’Italia, che conta oltre 4 milioni di disabili (si tratta di un valore stimato, poiché non ci sono dati certi a riguardo), non è un paese a misura di disabile per diverse ragioni tra cui l’esiguità dei fondi messi a disposizione, il clima discriminatorio, le barriere architettoniche, le disuguaglianze in campo occupazionale, sanitario e scolastico.
 Come colpisce questa patologia? La Sm evolve da una fase acuta infiammatoria iniziale ad una fase cronica, quindi, una volta insorta, sarà presente per sempre nella vita del paziente.
Come colpisce questa patologia? La Sm evolve da una fase acuta infiammatoria iniziale ad una fase cronica, quindi, una volta insorta, sarà presente per sempre nella vita del paziente. Nel 1869 il neurologo Jean Martin Charcot definì i sintomi clinici della malattia e i primi criteri diagnostici, raggruppati nella triade che da lui prende il nome: nistagmo, tremore intenzionale e parola scandita.
Nel 1869 il neurologo Jean Martin Charcot definì i sintomi clinici della malattia e i primi criteri diagnostici, raggruppati nella triade che da lui prende il nome: nistagmo, tremore intenzionale e parola scandita.
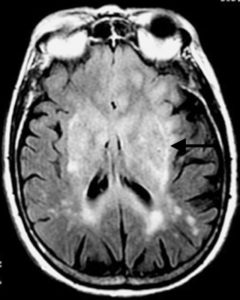 Le placche possono formarsi in ogni punto del Snc, ma sono più frequenti attorno ai ventricoli degli emisferi e del tronco cerebrale, nelle formazioni ottiche, nei peduncoli cerebellari superiori e medi e nel midollo spinale. Al microscopio ottico, le placche rosee si presentano con segni di infiammazione e sono localizzate prevalentemente in zona perivenulare, contengono linfociti T e plasmacellule mentre la mielina si presenta rigonfiata e incapace di assumere la tipica colorazione istologica di Niessel. Inoltre essa risulta frammentata e i suoi frammenti vengono fagocitati dai macrofagi. Le placche grigie sono croniche, in esse la mielina è andata persa e sostituita dalla deposizione di fibre prodotte dalle cellule connettivali, con conseguente cicatrizzazione o sclerosi, che porta allo stiramento e alla frammentazione degli assoni. Finché questi ultimi non sono lesionati, è possibile una riparazione perché la mielina può rigenerarsi. Quando però gli assoni si lesionano, diventano incapaci di trasmettere l’impulso nervoso. Quest’ultimo peraltro rallenta moltissimo già con la perdita del rivestimento di mielina.
Le placche possono formarsi in ogni punto del Snc, ma sono più frequenti attorno ai ventricoli degli emisferi e del tronco cerebrale, nelle formazioni ottiche, nei peduncoli cerebellari superiori e medi e nel midollo spinale. Al microscopio ottico, le placche rosee si presentano con segni di infiammazione e sono localizzate prevalentemente in zona perivenulare, contengono linfociti T e plasmacellule mentre la mielina si presenta rigonfiata e incapace di assumere la tipica colorazione istologica di Niessel. Inoltre essa risulta frammentata e i suoi frammenti vengono fagocitati dai macrofagi. Le placche grigie sono croniche, in esse la mielina è andata persa e sostituita dalla deposizione di fibre prodotte dalle cellule connettivali, con conseguente cicatrizzazione o sclerosi, che porta allo stiramento e alla frammentazione degli assoni. Finché questi ultimi non sono lesionati, è possibile una riparazione perché la mielina può rigenerarsi. Quando però gli assoni si lesionano, diventano incapaci di trasmettere l’impulso nervoso. Quest’ultimo peraltro rallenta moltissimo già con la perdita del rivestimento di mielina.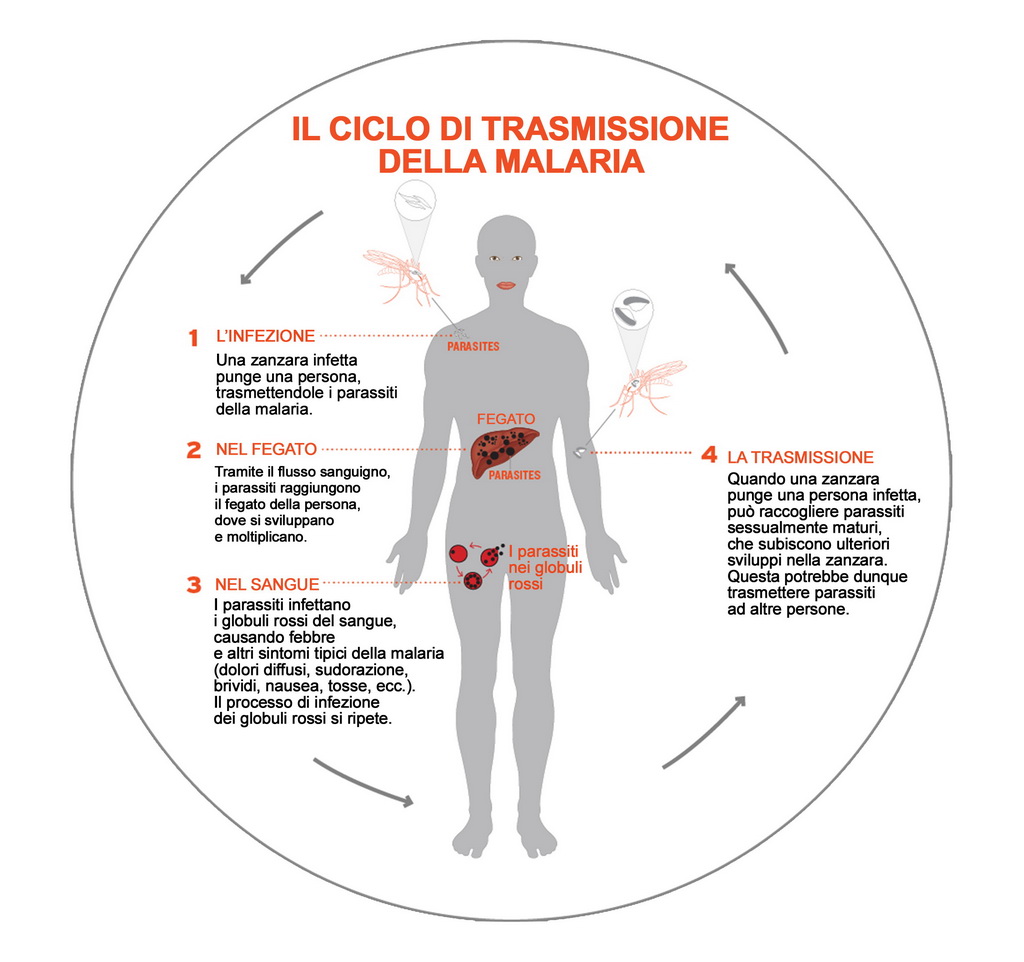
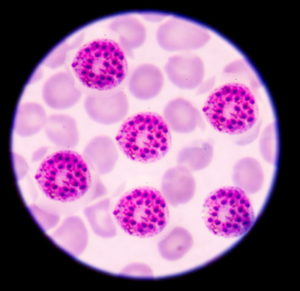 Il plasmodium iniettato dalla zanzara entra nei globuli rossi e comincia a crescere e dividersi aumentando in numero e volume finché non causa la rottura della membrana del globulo rosso infestando il sangue, lo stesso sangue che sarà risucchiato da altre zanzare e verrà rimesso nel sangue di qualche altra persona (grafico a pag. 64).
Il plasmodium iniettato dalla zanzara entra nei globuli rossi e comincia a crescere e dividersi aumentando in numero e volume finché non causa la rottura della membrana del globulo rosso infestando il sangue, lo stesso sangue che sarà risucchiato da altre zanzare e verrà rimesso nel sangue di qualche altra persona (grafico a pag. 64).
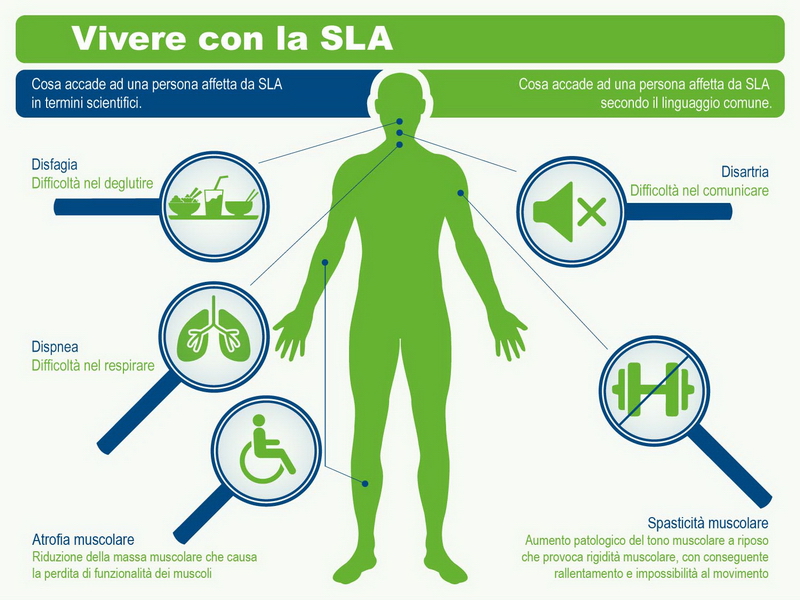


 Attualmente non esiste una cura efficace per la Sla. L’unico farmaco approvato dalla Fda (Federal drugs administration statunitense) in grado di rallentarne il decorso per qualche mese è il riluzolo, che in Italia viene somministrato solo a livello ospedaliero.
Attualmente non esiste una cura efficace per la Sla. L’unico farmaco approvato dalla Fda (Federal drugs administration statunitense) in grado di rallentarne il decorso per qualche mese è il riluzolo, che in Italia viene somministrato solo a livello ospedaliero.